
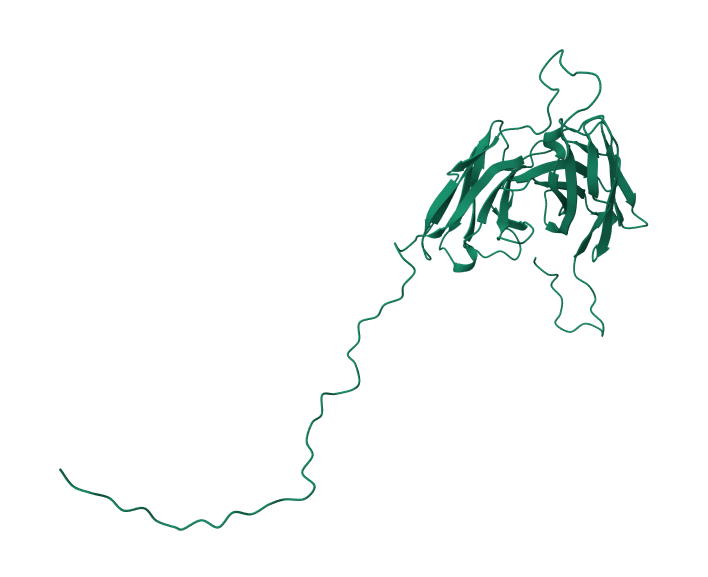
scFv Structure Modelling
A bit on Interleukin-8
Interleukin-8 (IL-8) plays a crucial role in protein binding by facilitating interactions with chemokine receptors, particularly CXCR1. IL-8 is a small globular protein with defined structural regions. It possesses binding sites primarily in its N-loop and 40s loop regions. These regions contain specific charged and hydrophobic residues that are essential for receptor recognition. When IL-8 encounters CXCR1, these binding sites on IL-8 engage with complementary sites on the receptor’s N-terminal domain, such as ND-CXCR1(1–38). These interactions create a stable protein-protein complex, which serves as a signaling mechanism to initiate cellular responses, such as chemotaxis and immune cell activation, ultimately aiding in immune responses and the regulation of inflammation.
This section lays out our process of predicting the structure for the anti-IL8 scFv format antibody. The amino acid sequence for the antibody has the following parts.
- A signal peptide sequence to ensure that the antibody is secreted from the cell.
MPLLLLLPLLWAGALA
- The variable heavy chain (VH) of the antibody.
EVQLLESGGGLVQPGGSLRLSCAASGFTFSYYGMGWVRQAPGKGLEWVSG
ISYSGSGTYYADSVKGRFTISRDNSKNTLYLQMNSLRAEDTAVYYCARDY
VGNLDYWGQGTLVTVSS
- A linker sequence to connect the VH and VL chains.
GGGGSGGGGSGGGGS
- The variable light chain (VL) of the antibody.
DIQMTQSPSSLSASVGDRVTITCRASQSISSYLNWYQQKPGKAPKLLIYA
ASSLQSGVPSRFSGSGSGTDFTLTISSLQPEDFATYYCQQSDTPSTFGQG
TKLEIK
- A 3xFLAG and a 6xHis-tag sequence to facilitate purification of the antibody.
RTDYKDHDGDYKDHDIDYKDDDDKAAALPETGGHHHHHH
Therefore, the full amino acid sequence that we will work with is
MPLLLLLPLLWAGALAEVQLLESGGGLVQPGGSLRLSCAASGFTFSYYGM
GWVRQAPGKGLEWVSGISYSGSGTYYADSVKGRFTISRDNSKNTLYLQMN
SLRAEDTAVYYCARDYVGNLDYWGQGTLVTVSSGGGGSGGGGSGGGGSDI
QMTQSPSSLSASVGDRVTITCRASQSISSYLNWYQQKPGKAPKLLIYAAS
SLQSGVPSRFSGSGSGTDFTLTISSLQPEDFATYYCQQSDTPSTFGQGTK
LEIKRTDYKDHDGDYKDHDIDYKDDDDKAAALPETGGHHHHHH
Note that we keep the signal peptide and the tags attached during the modelling process, because we do not know whether (and how) they interact with Interleukin-8 during docking.
Source
O-IL8-15 Biological Probe, in Structural Genomics Consortium: thesgc.org/biological-probes/il-8
AlphaFold2
AlphaFold2 is a deep learning system that predicts protein structures from amino acid sequences. We used the open-source distribution of AlphaFold2, ColabFold to predict the structure of the antibody. We used the AlphaFold2_mmseqs2 notebook. This notebook differs from full AlphaFold2 and AlphaFold2 Colab in that it uses MMseqs2 (Many-against-Many sequence searching) in place of homology detection and MSA pairing.
We used ColabFold with two different schemes: one without templates, and one with PDB70 as a database for templates. We also relaxed the top structure in either scheme with AMBER.
With PDB70
The structure generated with PDB70 is available on our github page: modelling section.

{kind=link}
Templates
The structure generated without templates is available on our github page: modelling section.

Data for analysis is present in the AlphaFold Without Template and AlphaFold with PDB70 directories, along with other generated models. Both of these have very similar average predicted aligned errors, as well as predicted lDDT scores. For both, folding is poor near the ends where the signal peptide and flags were attached, and in the middle
where the linker is present.
| With PDB70 | Without Templates |
 |  |
 |  |
 |  |
Alpha Fold Interpretation
PLDDT:
AlphaFold produces a per-residue estimate of its confidence on a scale from 0 – 100. This confidence measure is called pLDDT and corresponds to the model’s predicted score on the lDDT-Cα metric. It is stored in the B-factor fields of the mmCIF and PDB files available for download (although unlike a B-factor, higher pLDDT is better). pLDDT is also used to colour-code the residues of the model in the 3D structure viewer. The following rules of thumb provide guidance on the expected reliability of a given region:
- Regions with pLDDT > 90 are expected to be modelled to high accuracy. These should be suitable for any application that benefits from high accuracy (e.g. characterising binding sites).
- Regions with pLDDT between 70 and 90 are expected to be modelled well (a generally good backbone prediction).
- Regions with pLDDT between 50 and 70 are low confidence and should be treated with caution.
- The 3D coordinates of regions with pLDDT < 50 often have a ribbon-like appearance and should not be interpreted. We show in our paper that pLDDT < 50 is a reasonably strong predictor of disorder, i.e. it suggests such a region is either unstructured in physiological conditions or only structured as part of a complex.
- Structured domains with many inter-residue contacts are likely to be more reliable than extended linkers or isolated long helices.
- Unphysical bond lengths and clashes do not usually appear in confident regions. Any part of a structure with several of these should be disregarded.
The pLDDT per position is also given as a plot for the five models made in every run and gives a simpler overview:

In the above graph we see prediction models of a protein presented in pLDDT graph. Most of the graph has pLDDT score above 75-80 and hence it has a high confidence level on the structure and relative inter atomic distances. Different models have different pLDDT scores and Model 1 has on average the highest level. Hence model 1 is the best predicted Structure.
PAE GRAPHS
Our protein clearly has 2 domains. We use the Predicted Aligned Error (PAE) plot provided by AlphaFold. PAE is a 2D plot.
The colour at (x, y) corresponds to the expected distance error in residue x’s position, when the prediction and true structure are aligned on residue y. Dark Blue is good (low error), red is bad (high error). For example, aligning on residue 150:
- We’re confident in the relative position of residue 100
- We’re not confident in the relative position of residue 200

Of course, the two low-error squares correspond to the two domains.
AlphaFold produces a per-residue confidence score (pLDDT) between 0 and 100. Some regions with low pLDDT may be unstructured in isolation.
Dark blue- Very High pLDDT (MORE CONFIDENCE)
Light Blue-Confident (pLDDT level moderate)
White-red- Low (pLDDT level low)
Red- Very low (pLDDT levels very low)
Database of Alpha Fold:
- 365 K predicted for proteins from 21 model organisms.
- For the organisms currently covered, predicted structures are available for the sequences in the UniProt reference proteome that are between 16 and 2700 amino acids long and contains only standard amino acids.
They use mmCIF files from the model archive extension to get resources and information of predicted proteins. It contains molecular description, Taxonomy id, Quality measures, per residue quality.
Impact of structural bioinformatics:
- Predicting complexes between macromolecules, about intrinsically disordered proteins and structures of protein-protein, protein-nuclei acid complexes.
- Provide information on protein dynamics i.e. relevant confirmation states.
- Ligand Predictions.
- It will accelerate Structural Biology. The structural studies and its uses in mechanising reactions of bio molecules.

MSA
{kind=link}
MSA: Multiple sequence alignment. Alpha fold predicts various possible structures for a given sequence of amino acids. So, one of the tools developed are the MSA graphs. Here we can take two different alignments and combine them based on requirements, organism and other information. We can pair and unpair them to get better results, which depends on the sequences and how well the software predicts on each of them.
Here we can see for unpaired MSA case, we have possible sequence counts and their positions and based on it, the software well developed 1 graph which has minimum error in relative positions. While in the case of Paired MSA, we have mostly all better than Unpaired case, but their quality is decreasing.
Thus combining proteins to form a bigger one can help us determine structures and relative inter atomic distances of that protein better.
The MSA help us to even determine structural accuracy of bigger proteins. For example, we want to determine the PAE graph for a protein P which is made of 2 copies of protein A, 1 copy of B and 2 copies of C, we can create MSA sequence of each and using the graphs we can create multiple possible structure of P with respective PAE graphs and choose the best one. Hence this simplifies and increase information regarding protein analysis.
Citations
- Mirdita, M., Schütze, K., Moriwaki, Y., Heo, L., Ovchinnikov, S., & Steinegger, M. (2022). ColabFold: Making Protein folding accessible to all. Nature Methods.
- Mirdita, M., Steinegger, M., & S”oding, J. (2019). MMseqs2 desktop and local web server app for fast, interactive sequence searches. Bioinformatics, 35(16), 2856–2858.
- Mirdita, M., Driesch, L., Galiez, C., Martin, M., S”oding, J., & Steinegger, M. (2017). Uniclust databases of clustered and deeply annotated protein sequences and alignments. Nucleic Acids Res., 45(D1), D170–D176.
- Mitchell, A., Almeida, A., Beracochea, M., Boland, M., Burgin, J., Cochrane, G., Crusoe, M., Kale, V., Potter, S., Richardson, L., Sakharova, E., Scheremetjew, M., Korobeynikov, A., Shlemov, A., Kunyavskaya, O., Lapidus, A., & Finn, R. (2019). MGnify: the microbiome analysis resource in 2020. Nucleic Acids Res..
- Steinegger, M., Meier, M., Mirdita, M., V”ohringer, H., Haunsberger, S., & S”oding, J. (2019). HH-suite3 for fast remote homology detection and deep protein annotation. BMC Bioinform., 20(1), 473.
- Berman, H., Henrick, K., & Nakamura, H.. (2003). Announcing the worldwide Protein Data Bank.
- Eastman, P., Swails, J., Chodera, J., McGibbon, R., Zhao, Y., Beauchamp, K., Wang, L.P., Simmonett, A., Harrigan, M., Stern, C., Wiewiora, R., Brooks, B., & Pande, V. (2017). OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLOS Comput. Biol., 13(7).
SWISS-MODEL
Model Building
The SWISS-MODEL server employs ProMod3, an in-house comparative modeling engine based on OpenStructure, to compute models. It utilizes template structures, statistical potentials, conformational searches, rotamer libraries, and energy minimization to generate accurate protein models, with computations powered by the OpenMM library and CHARMM22/CMAP force field.
Modelling Modes
Depending on the difficulty of the modelling task, three different types of modelling modes are provided, which differ in the amount of user intervention: automated mode, alignment mode, and project mode.
Automated Mode:
The Automated Mode in SWISS-MODEL requires only the protein’s amino acid sequence or UniProtKB accession code as input. It employs BLAST and HHblits to identify templates, ranks them by model quality, and automatically selects multiple templates if they cover distinct protein regions or represent alternative conformations.
Alignment Mode:
If the desired template for modelling is known and available in the SWISS-MODEL Template Library (SMTL), a target–template alignment in either FASTA or Clustal format may be used to start the modelling process, thereby skipping the template search.
The template sequence(s) should be named using the PDB ID format (i.e. “1CNR” or “1CNR_A”). The user will be asked to specify which sequence in the alignment corresponds to the target and/or the template protein from a drop-down list. The Alignment mode allows the advanced user to invoke the modelling step starting from alternative alignments and to evaluate the quality of these alternative models.

{kind=link}
Project Mode:
In complex modeling scenarios, manual alignment adjustment is valuable when sequence-based methods are inconclusive. The DeepView – Swiss-PdbViewer program, accessible in SWISS-MODEL, empowers users to generate, view, and fine-tune modeling projects by adjusting template structures, alignments, and insertions/deletions in 3D structures.
The SWISS-MODEL Template Library (SMTL)
The SWISS-MODEL template library, derived from the Protein Data Bank (PDB), is a pivotal resource for the modeling pipeline. It houses experimentally determined protein structures and offers atomic coordinates, along with searchable sequence and profile databases using BLAST and HHblits. Importantly, it provides alignment-independent features such as residue mapping, predicted solvent accessibility, and secondary structure data. Users can interactively explore entries, and ligands are classified as synthetic, natural, or part of crystallization buffers, aiding the decision-making process for their inclusion in the final protein models.

{kind=link}
Input Data
The amino acid sequence of the target protein can be submitted either as plain text, or in FASTA format.
Example of plain text sequence:

Example of FASTA sequence:

Display of modelling results
This section offers a 3D visualization of models and their target-template sequence alignment, along with the option to download model coordinates. Various sequence features and scoring schemes are synchronized with the 3D view. Users can customize alignment coloring via the “Options” button. Models are available in DeepView project files or PDB format. Information on selected templates is linked to the SWISS-MODEL Template library. Models are displayed with QMEAN-based coloring for assessing quality, with additional details on oligomeric state, ligands, and cofactors. Users can opt for a formatted report page or download an archive with all models and reports for the target sequence.

{kind=link}
Model evaluation
Global Model Evaluation:
GMQE and QMEANDisCo Global Scores: These metrics provide an overall assessment of model quality, ranging from 0 to 1, with higher values indicating better quality. GMQE considers coverage, meaning a model covering only half the target sequence won’t score above 0.5, while QMEANDisCo evaluates models without coverage dependency. GMQE (Global Model Quality Estimate): GMQE combines properties from the target-template alignment and template structure. It employs a multilayer perceptron to predict the lDDT score. GMQE aids template selection by providing quality estimates before model creation. After building a model, GMQE updates based on the QMEANDisCo score, enhancing quality estimation. When using AlphaFold DB templates, GMQE sums per-residue plDDT values of aligned template residues normalized by target sequence length.
QMEANDisCo Global Score: This score averages per-residue QMEANDisCo scores, known to correlate with lDDT scores. The provided error estimate reflects the standard deviation between QMEANDisCo global scores and lDDT scores for models of similar size.
QMEANDisCo is not calculated for models using AlphaFold DB templates.
QMEAN Z-score Analysis: This deprecated analysis used four statistical potentials of mean force to calculate the “QMEAN” score and Z-scores. A Z-score close to 0.0 indicates a native-like structure, while a Z-score below -4.0 suggests low-quality models. The “Comparison” plot displays protein length on the x-axis and the “QMEAN” score on the y-axis. Experimental structures falling within 1 standard deviation are black, those within 1-2 standard deviations are grey, and structures further from the mean are light grey. The model is represented as a red star. QMEAN Z-score analysis is not performed for models using AlphaFold DB templates.
In summary, GMQE and QMEANDisCo Global Scores offer comprehensive quality assessments for protein models, considering alignment, template structure, and statistical correlations. They replace the deprecated QMEAN Z-score analysis for more reliable model quality estimation.
SWISS-MODEL is a fully automated protein structure homology-modelling tool. SWISS-MODEL searches for templates against the SWISS-MODEL template library (STML). The list of suggested templates can be found on our github page: modelling section. The top 2 templates, in order of expected quality (GMQE score), are:
- AlphaFold DB model of Q65ZC9_HUMAN (Q65ZC9.1.A)
- Crystal Structure of spFv GLK1 HL (8dy0.1.A)
We used both these templates to generate structures for the antibody, since they are obtained from drastically different paradigms–the former from deep learning, and the latter from X-ray crystallography. Both have nearly the same sequence identity with the antibody, as well as GMQE scores.
Q65ZC9.1.A
The structure generated from the AlphaFold DB model of Q65ZC9_HUMAN is available at https://github.com/iGEMIISc/scFv-Modelling/blob/main/SWISS%20MODEL/models/01/model.pdb

8dy0.1.A
The structure generated from the Crystal Structure of scFv GLK1 HL is available at
https://github.com/iGEMIISc/scFv-Modelling/blob/main/SWISS%20MODEL/models/02/model.pdb

More details about the models can be found in the SWISS MODEL directory.
Citations
- Waterhouse, A., Bertoni, M., Bienert, S., Studer, G., Tauriello, G., Gumienny, R., Heer, F.T., de Beer, T.A.P., Rempfer, C., Bordoli, L., Lepore, R., Schwede, T. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46(W1), W296-W303 (2018).
- Bienert, S., Waterhouse, A., de Beer, T.A.P., Tauriello, G., Studer, G., Bordoli, L., Schwede, T. The SWISS-MODEL Repository – new features and functionality. Nucleic Acids Res. 45, D313-D319 (2017).
- Studer, G., Tauriello, G., Bienert, S., Biasini, M., Johner, N., Schwede, T. ProMod3 – A versatile homology modelling toolbox. PLOS Comp. Biol. 17(1), e1008667 (2021).
- Studer, G., Rempfer, C., Waterhouse, A.M., Gumienny, G., Haas, J., Schwede, T. QMEANDisCo – distance constraints applied on model quality estimation. Bioinformatics 36, 1765-1771 (2020).
- Bertoni, M., Kiefer, F., Biasini, M., Bordoli, L., Schwede, T. Modeling protein quaternary structure of homo- and hetero-oligomers beyond binary interactions by homology. Scientific Reports 7 (2017).
I-TASSER
I-TASSER (Iterative Threading ASSEmbly Refinement) is a protein structure prediction server, that identifies structural templates from the PDB by multiple threading approach (LOMETS), and constructs structures by iterative template-based fragment assembly simulations. I-TASSER used Crystal structure of SARS spike protein receptor, 2ghw as the template in its more reliable structure.

Other templates used by I-TASSER were also proteins whose structures are known through experimental techniques like X-ray diffraction or electron microscopy. More details about these models can be found in the I-TASSER directory.
Citations
- Wei Zheng, Chengxin Zhang, Yang Li, Robin Pearce, Eric W. Bell, Yang Zhang. Folding non-homology proteins by coupling deep-learning contact maps with I-TASSER assembly simulations. Cell Reports Methods, 1: 100014 (2021).
- Chengxin Zhang, Peter L. Freddolino, and Yang Zhang. COFACTOR: improved protein function prediction by combining structure, sequence and protein-protein interaction information. Nucleic Acids Research, 45: W291-299 (2017).
- Jianyi Yang, Yang Zhang. I-TASSER server: new development for protein structure and function predictions, Nucleic Acids Research, 43: W174-W181, 2015.
Modeller
Modeller is a software package for protein structure modelling. We will use Modeller to generate structures for the antibody using templates obtained from querying PDB. We identified 6Y6C: Entity 2 from the PDB as the top template for our antibody. This protein is an extracellular domain with an scFv-fragment. This template had a sequence identity of 93% with our antibody, and an E-value of 1.087e-135.
Chain-C of 6Y6C aligned the best with our target sequence, which we determined using Modeller. We then used Modeller to produce the structure for our anti-body using this chain. We ran the structure through energy-optimization as well, but it seemed that the structure was already optimized since therewas no change in its DOPE score
More details about the modelling process can be found in the Modeller directory.
Citations
- B. Webb, A. Sali. Comparative Protein Structure Modeling Using Modeller. Current Protocols in Bioinformatics 54, John Wiley & Sons, Inc., 5.6.1-5.6.37, 2016.
- M.A. Marti-Renom, A. Stuart, A. Fiser, R. Sánchez, F. Melo, A. Sali. Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 29, 291-325, 2000.
- A. Sali & T.L. Blundell. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779-815, 1993.
- A. Fiser, R.K. Do, & A. Sali. Modeling of loops in protein structures, Protein Science 9. 1753-1773, 2000.
Comparative Analysis with ProDy
We select the 6 models discussed above for comparative analysis. All data in this section has been generated using ProDy. The number of resolved atoms in given below.
| Model | Number of Resolved Atoms |
| AlphaFold2 with PDB70 | 4281 |
| AlphaFold2 without Templates | 4281 |
| SWISS-MODEL Q65ZC9.1.A | 1769 |
| SWISS-MODEL 8dy0.1.A | 1733 |
| I-TASSER | 4277 |
| Modeller | 2201 model |
{kind=link}
This immediately places the I-TASSER model in a much more reliable position than models generated by SWISS-MODEL and Modeller. In fact, it gets quite close to the coverage of AlphaFold2 models which were AI-generated.
Root Mean Square Deviation (RMSD)
We now see the RMSD of the AlphaFold2 and I-TASSER models with respect to each other. This is done after superimposing the models on the same reference structure, through the Kabsch algorithm.
| AlphaFold2 with PDB70 | AlphaFold2 without Templates | I-TASSER | |
| AlphaFold2 with PDB70 | 0 | 1.53 | 12.17 |
| AlphaFold2 without Templates | 1.53 | 0 | 12.57 |
| I-TASSER | 12.17 | 12.57 | 0 |
The AlphaFold2 model generated with PDB70 therefore appears to be the most reliable, since it is the closest to other structures.
Citations
- Zhang S, Krieger JM, Zhang Y, Kaya C, Kaynak B, Mikulska-Ruminska K, Doruker P, Li H, Bahar I ProDy 2.0: Increased scale and scope after 10 years of protein dynamics modelling with Python 2021 Bioinformatics, btab187.
- Bakan A, Meireles LM, Bahar I ProDy: Protein Dynamics Inferred from Theory and Experiments 2011 Bioinformatics 27(11):1575-1577
- Bakan A, Dutta A, Mao W, Liu Y, Chennubhotla C, Lezon TR, Bahar I Evol and ProDy for Bridging Protein Sequence Evolution and Structural Dynamics 2014 Bioinformatics 30(18):2681-2683
Docking of anti-IL8 scFv to IL8
Note on protein models
The model for the anti-IL8 scFv (anti-interleukin-8) (single-chain variable fragment) is taken from our scFv-Modelling repository. In particular, we have chosen the ColabFold model generated with PDB70. We have used ColabFold for a graphic processing unit for folding proteins in a 3-Dimensional format. It has a GDT_TS (Global distance Test) score of 92.4% . This is a rating of the comparision of protein structures with well-known amino acid sequences. ColabFold performs a MMseqs-2 based homology search server to find the template of the protein.

Interleukin 8 (IL-8) is a chemokine produced by various cell types. This acts as a mediator of inflammation in the tissues and a potent angiogenic factor. It attracts various kinds of phagocytic cells such as neutrophils, basophils and T-cells to stimulate the phagocytosis of cells and promote angiogenesis. Being a chemokine, it promotes the immune cell migration for the phagocytosis. These belong to a set of monoclonal antibodies which have become a modern class of medicines used for treating various diseases such as cancer, cardiovascular and inflammation diseases. Interleukin 8 is in culture a 72 amino acid peptide in it’s major form when it is secreted by macrophages. The scFv antibody fragments consist of variable regions of heavy (VH) and light chain (VL) of full antibodies that are joined by a linker sequence.

The model for IL8 is taken from the PDB (PDB ID: 2IL8). This PDB file consists of 30 models, each with a different conformation of IL8 in a 3D structure. We have chosen the first model for our docking. The PDB library consists of the 3-Dimensional structure of the given protein and all the sequences and mutations of the fragments of the protein. The solution NMR method has been used to experimentally verify the protein structure and so far 30 different conformers of the same molecule have been shown to exist. We use a homology based modeling pattern for this protein which consists of building the given protein based on the atomic resolution models of the constituent amino acids sequences.
Global Range Molecular Matching (GRAMM)
GRAMM is a docking web server that maps the intermolecular energy landscape by predicting a spectrum of docking poses corresponding to stable (deep energy minima) and transient (shallow minima) protein interactions. This program performs an exhaustive comparision in 6-Dimensional structures through the relative translations and rotations of the molecule. It performs a comprehensive comparision in which it accounts for all the possible orientations and positions of the molecule in study against another known molecule. It simulates the way in which atoms interact with each other when they are at various distances from each other and predict the pose in which the molecule attains the most energitically favourable state when the two interact with each other. It tests against all the available models in the PDB and gives an exhaustive result.
GRAMM does not perform a statistical sampling of the model but produces an exhaustive search against all configurations of the complex and give the one with the best steric fit score.
We used the anti-IL8 scFv as the receptor and IL8 as the ligand.

Free Docking
In a free docking algorithm, many conformations of the two proteins are evaluated to minimize the energy of their interaction. It is a simulation of the behaviour and interaction of the molecule and identification of the potential binding sites of the molecule.
Steps involved in the process of free docking:
- Preparing the ligand and receptor structures, which may require removing water molecules, adding hydrogen atoms, assigning charges, and defining flexible regions. This is done to prepare the molecule for all possible sites of binding.
- Generating possible poses of the ligand in the binding site of the receptor, which may involve sampling different orientations, conformations, and translations of the ligand.
- Evaluating the poses based on their energy and/or scoring function, which may include terms for electrostatics, van der Waals, solvation, entropy, and empirical factors. This gives the best configuration of the most stable energy state.
- Ranking the poses according to their score and selecting the best ones for further analysis or validation.

Template-based Docking
In the template-based method, a search is made from the PDB to identify heterodimer templates for our target. Then, homology modelling is used to map the target sequence onto the template structure. This is used to generate template-based structural models of assemblies using close and remote homologs of known 3D structures which are detected through an automated search protocol.

The structural modelling of protein interactions in the absence of close homologous templates is a challenging task. Recently, template-based docking methods have emerged to exploit local structural similarities to help ab-initio protocols provide reliable 3D models for protein interactions. To efficiently utilize available homologous complexes in the protein data bank (PDB) (24), we have developed a hybrid docking strategy to automatically incorporate the binding interface information into traditional global docking.
We will be taking the top models from both the free docking and template-based docking methods for further analysis. It should be noted that these cannot be directly compared as the free docking method results are expressed in terms of shape complementarity while the template-based docking method results are expressed in terms of AACE18 (atomic contact energy) scores. This potential determines the relation between the binding free energy and SASA (Solvent Accessible Surface Area) values.
AutoDock-
AutoDock3, a suite of open-source software for docking and virtual screening. AutoDock consists of several programs, such as AutoDock4, which performs the docking calculations using a Lamarckian genetic algorithm and a scoring function that accounts for torsional entropy; AutoGrid4, which pre-calculates the grid maps for the receptor; and AutoDockTools, which provides a graphical user interface for preparing and analyzing the docking inputs and outputs.
AutoDock hasn’t been utilized by us for this protein modelling as it does not provide for appropriate calculations for large molecules of ligands that we have used. It is unable to account for flexibility of cyclic and macro-cyclic ligands. It also requires much higher computational and graphical power for running the model which could amount to a significant cost of operation for a large ligand molecule like ours. Also, it often leads to unreliable results when small molecules are docked into flexible binding sites. Since our protein contains a larger sequence (72 in general and 8452 Da) of amino acid structure, it is not suitable to use AutoDock directly for docking our protein.
Other molecular docking softwares:
- SwissDock
- Mcule
For our simulation of the binding energy, we have used PRODIGY (PROtein binDIng enerGY prediction) software for all the docking and calculations. It has the following features:
- Predicts the binding affinity in PROTEIN-PROTEIN complexes.
- Predicts the binding affinity in PROTEIN-SMALL LIGAND complexes.
- Classification interfaces between biological or crystallographic
By default, all intermolecular contacts are taken into consideration, a molecule being defined as an isolated group of amino acids sharing a common chain identifier. In specific cases, for example antibody-antigen complexes, some chains should be considered as a single molecule. The PRODIGY server implements our simple but highly effective predictive model based on intermolecular contacts and properties derived from non-interface surface.

Citations
- Katchalski-Katzir, E., Shariv, I., Eisenstein, M., Friesem, A.A., Aflalo, C., Vakser, I.A., 1992, Molecular surface recognition: Determination of geometric fit between proteins and their ligands by correlation techniques, Proc. Natl. Acad. Sci. USA, 89:2195-2199.
- Vakser, I.A., 1996, Long-distance potentials: An approach to the multiple-minima problem in ligand-receptor interaction, Protein Eng., 9:37-41.
- Porter, K. A., Desta, I., Kozakov, D., & Vajda, S. (2019). What method to use for protein–protein docking? Current Opinion in Structural Biology, 55, 1–7.
- Rosell M., Fernández-Recio J. Docking approaches for modeling multi-molecular assemblies. Curr. Opin. Struct. Biol. 2020; 64:59–65.
- Waterhouse A., Bertoni M., Bienert S., Studer G., Tauriello G., Gumienny R., Heer F.T., de Beer T.A.P., Rempfer C., Bordoli L. et al. . SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018; 46:W296–W303.
Analysis of Docking Results using PRODIGY
PRODIGY (PROtein binDIng enerGY prediction) is a web server for predicting the binding affinity of protein-protein complexes. We used this tool to analyze the docking results obtained from GRAMM. To analyze the binding affinity, the PRODIGY webserver was employed where the binding affinity within the protein-protein complex is predicted based on contact-based methods. It performs binding affinity analysis based on the calculation of Gibbs free energy (∆G| kcal mol-1 ) and dissociation constant (Kd | M).
| Method | Predicted Binding Affinity ΔG in kcal mol−1 | Predicted Dissociation Constant Kd in M at 36°C |
| Free Docking | −16.5 | 2.2×10−12 |
| Template-based Docking | −7.4 | 6.3×10−6 |
The calculations are done for 36°C as this is the temperature at which our antibody will interact with IL8 in the human peritoneum*. It can be seen that free docking has provided a much more favourable binding affinity and dissociation constant than template-based docking. This suggests that the free docking could access a more stable conformation of the complex, and thus is more likely to be closer to the actual binding conformation.
Detailed results are available in the PRODIGY directory.
Citations
- Vangone A. and Bonvin A.M.J.J. “Contact-based prediction of binding affinity in protein-protein complexes”, eLife, 4, e07454 (2015).
- Xue L., Rodrigues J., Kastritis P., Bonvin A.M.J.J., Vangone A., “PRODIGY: a web-server for predicting the binding affinity in protein-protein complexes”, Bioinformatics, doi:10.1093/bioinformatics/btw514 (2016).
- Andrusier N, Mashiach E, Nussinov R, Wolfson HJ (2008) Principles of flexible protein-protein docking. Proteins 73:271–289
- Karaca E, Bonvin AMJJ (2013) Advances in integrative modeling of biomolecular complexes. Methods 59:372–381
Other References
- *Ott, D.E. The peritoneum and the pneumoperitoneum: a review to improve clinical outcome. Gynecol Surg 1, 101–106 (2004). https://doi.org/10.1007/s10397-004-0019-y
- Study showing that GRAMM can successfully predict protein-protein interactions even for models of varying accuracy.
- Kundrotas P.J. et al. (2018) Modeling CAPRI targets 110–120 by template-based and free docking using contact potential and combined scoring function. Proteins, 86, 302–310. Study indicating the effectiveness of GRAMM and ranking it among the top performers in some CAPRI rounds.
- Study indicating methods and tools used for protein-protein docking.
Credits
We would like to thank Amar Singh from the Center for Computational Biology at the University of Kansas for answering our queries regarding the use of GRAMM, particularly with respect to the evaluation of the docking results.
Docking-aptide-fibronectin
The protein models:
The model for the aptide is taken from PDB (PDB ID: 2MNU, Chain B). The PDB file was split and only the Chain B corresponding to APT_EDB was taken.
The model for the fibronectin extra-domain B is taken from PDB (PDB ID: 2FNB).
Global Range Molecular Matching (GRAMM)
GRAMM is a docking web server that maps the intermolecular energy landscape by predicting a spectrum of docking poses corresponding to stable (deep energy minima) and transient (shallow minima) protein interactions. We used the Fibronectin extradomain-B (PDB ID: 2MNU) as the receptor and the aptide APT_EDB (PDB ID: 2MNU, Chain B) as the ligand.
Free Docking
In a free docking algorithm, a large number of conformations of the two proteins are evaluated to minimize the energy of their interaction.
Template-based Docking
In the template-based method, a search is made from the PDB to identify heterodimer templates for our target. Then, homology modelling is used to map the target sequence onto the template structure.
We will be taking the top models from both the free docking and template-based docking methods for further analysis. It should be noted that these cannot be directly compared as the free docking method results are expressed in terms of shape complementarity while the template-based docking method results are expressed in terms of AACE18 (atomic contact energy) scores.
Analysis of Docking Results using PRODIGY
PRODIGY (PROtein binDIng enerGY prediction) is a web server for predicting the binding affinity of protein-protein complexes. We used this tool to analyze the docking results obtained from GRAMM.
| Method | Predicted Binding Affinity ΔG0 in kcal mol−1 | Predicted Dissociation Constant Kd in M at 36°C |
| Free Docking | −10.0 | 7.9×10−8 |
| Template-based Docking | −6.8 | 3.8×10−5 |
The calculations are done for 36°C as this is the temperature at which our lipid nanoparticle decorated with aptides will interact with extra-domain B of fibronectin in the human peritoneum*. It can be seen that free docking has provided a much more favourable binding affinity and dissociation constant than template-based docking. This suggests that the free docking could access a more stable conformation of the complex, and thus is more likely to be closer to the actual binding conformation.
The results obtained from docking can be found in the results section.
Toxicity Estimation of Components of our mRNA-LNP:
Toxicity Estimation of our Anti-IL8:
We utilise our amino acid sequence to estimate the toxicity of our anti-IL8 protein:
MPLLLLLPLLWAGALAEVQLLESGGGLVQPGGSLRLSCAASGFTFSYYGM
GWVRQAPGKGLEWVSGISYSGSGTYYADSVKGRFTISRDNSKNTLYLQMN
SLRAEDTAVYYCARDYVGNLDYWGQGTLVTVSSGGGGSGGGGSGGGGSDI
QMTQSPSSLSASVGDRVTITCRASQSISSYLNWYQQKPGKAPKLLIYAAS
SLQSGVPSRFSGSGSGTDFTLTISSLQPEDFATYYCQQSDTPSTFGQGTK
LEIKRTDYKDHDGDYKDHDIDYKDDDDKAAALPETGGHHHHHH
To check for toxicity, we utilize CSM-Toxin, a web server for predicting protein toxicity, developed by Biosig Lab. CSM-Toxin provides a comprehensive suite for rapid identification of toxin proteins. Having uploaded our AA sequence to the server, the following result was obtained:
| ID | Sequence | Prediction |
| PLLLLLPLLWAGALAEVQLLESGGGLVQPGGSLRLSCAASGFTFSYYGM | GWVRQAPGKGLEWVSGISYSGSGTYYADSVKGRFTISRDNSKNTLYLQMNSLRAEDTAVYYCARDYVGNLDYWGQGTLVTVSSGGGGSGGGGSGGGGSDIQMTQSPSSLSASVGDRVTITCRASQSISSYLNWYQQKPGKAPKLLIYAASSLQSGVPSRFSGSGSGTDFTLTISSLQPEDFATYYCQQSDTPSTFGQGTKLEIKRTDYKDHDGDYKDHDIDYKDDDDKAAALPETGGHHHHHH | Non-toxic |
A detailed CSV was also provided as output, along with the following graph.

Where a higher value indicates more toxicity of a given attention head. Clearly, our protein had close to no toxicity.
How it works:
This study used UniProt release 2022_04 data to identify toxic and non-toxic proteins. The model was trained using CD-HIT version 4.8.1, discarding sequences with non-standard residue codes. The final dataset contains 2475 toxic sequences and 214,740 non-toxic sequences, with a toxic to non-toxic ratio of approximately 90. The predictive model was built using raw amino acid sequences without additional features extracted or generated.
The curated sequences were divided into two groups: 203 toxic and 2337 non-toxic sequences uploaded after July 2021, used in a blind test set for CSM-Toxin and ToxinPred2, and 236 positive and 21,294 negative sequences for cross-validation. The remaining data was split into five parts for training and validation, with average performance metrics.
ProteinBERT was used as a base of the model. ProteinBERT, a model inspired by BERT, treats amino acids as words and protein sequences as sentences. It uses attention mechanisms to capture complex connections between amino acids. ProteinBERT was pre-trained using the Masked Language Model technique, capturing connections between amino acids and their surroundings. The model was pre-trained on over 100 million protein sequences from the UniProt database. The focus was on Global Ontology for binary prediction (toxic/non-toxic) based on the entire sequence.
Toxicity of various components of our lipid.
Our lipid composition is as follows:
- DSPC: 790.15 g/mol
- ALC-0315: 766.29 g/mol
- Cholesterol: 386.65 g/mol
- PEG: 2941.642 g/mol
To determine toxicity of individual components, we utilised the Toxicity Estimation Software Tool (TEST). Out of the available endpoints present in the software, we used
- The Oral Rat LD50 Endpoint: This indicates a concentration of a given chemical that leads to the death of 50% rats in a given population when administered orally.
| Query | SmilesRan | Exp_Value: -Log10(mol/kg) | Pred_Value: -Log10(mol/kg) | Exp_Value: mg/kg | Pred_Value: mg/kg |
| PEG | [H]OCCO | 1.12 | 1.89 | 4698.42 | 802.86 |
| DSPC | O=C(OCC(OC(=O)CCCCCCCCCCCCCCCCC)COP(=O)([O-])OCC[N+](C)(C)C)CCCCCCCCCCCCCCCCC | N/A | N/A | N/A | N/A |
| Cholesterol | [H]C12CC=C3CC(O)CCC3(C)C2([H])CCC4(C)C([H])(CCC14[H])C(C)CCCC(C)C | N/A | 2.55 | N/A | 1078.24 |
| ALC-0315 | O=C(OCCCCCCN(CCCCO)CCCCCCOC(=O)C(CCCCCC)CCCCCCCC)C(CCCCCC)CCCCCCCC | N/A | N/A | N/A | N/A |
We further observe that neither experimental nor predicted values of toxic concentrations were present for DSPC AND ALC-0315. Hence, we further utilise the software’s ability to provide results for similar chemicals which generated the following data:
Predictions for the test chemical and for the most similar chemicals in the training set for DSPC:


Predictions for the test chemical and for the most similar chemicals in the training set for ALC-0315:


- Ames Mutagenicity: This checks if the compound is positive for mutagenicity if it induces revertant colony growth in ant strain of Salmonella typhimurium.
| Query | SmilesRan | Exp_Value: -Log10(mol/kg) | Pred_Value: -Log10(mol/kg) | Exp_Result: | Pred_Result: mg/kg |
| DSPC | [O=C(OCC(OC(=O)CCCCCCCCCCCCCCCCC)COP(=O)([O-])OCC[N+](C)(C)C)CCCCCCCCCCCCCCCCC | N/A | -0.01 | N/A | Mutagenicity Negative |
| Cholesterol | [H]C12CC=C3CC(O)CCC3(C)C2([H])CCC4(C)C([H])(CCC14[H])C(C)CCCC(C)C | N/A | 0.2 | N/A | Mutagenicity Negative |
| PEG | [[H]OCCO | 0.00 | -0.03 | Mutagenicity Negative | Mutagenicity Negative |
| ALC-0315 | O=C(OCCCCCCN(CCCCO)CCCCCCOC(=O)C(CCCCCC)CCCCCCCC)C(CCCCCC)CCCCCCCC | N/A | 0.03 | N/A | Mutagenicity Negative |
We observe that all the compounds in our batch are found to be Mutagenicity Negative.
- Developmental Toxicity: This checks whether a chemical causes developmental toxicity effects to humans or animals.
| Query | SmilesRan | Exp_Value: -Log10(mol/kg) | Pred_Value: -Log10(mol/kg) | Exp_Result: | Pred_Result: mg/kg |
| DSPC | O=C(OCC(OC(=O)CCCCCCCCCCCCCCCCC)COP(=O)([O-])OCC[N+](C)(C)C)CCCCCCCCCCCCCCCCC | N/A | N/A | N/A | N/A |
| Cholesterol | H]C12CC=C3CC(O)CCC3(C)C2([H])CCC4(C)C([H])(CCC14[H])C(C)CCCC(C)C | N/A | 0.97 | N/A | Developmental Toxicant |
| PEG | [H]OCCO | N/A | 0.28 | N/A | Developmental NON-Toxicant |
| ALC-0315 | O=C(OCCCCCCN(CCCCO)CCCCCCOC(=O)C(CCCCCC)CCCCCCCC)C(CCCCCC)CCCCCCCC | N/A | N/A | N/A | N/A |
We observe that some of the above compounds in the batch can be toxic to animals like Cholesterol.
In single chemical mode, one can generate predicted environmental transformation products by checking “Run CTS” which can help us create suitable environment like hydrolysis, abiotic reduction and human metabolism. These tests have been made to consider “human metabolism” in animal cells. If likely transformation products are generated, the toxicity will be displayed.
This test is especially useful as we are using the mRNA on animal cells, so this will be able to tell us to what extent Cholesterol and other compounds can be used to avoid toxicity.
Issues with the method:
Clearly, while TEST works well for some of the chemicals present in our LNP, we observe that the first major issue is the metric of testing toxicity of chemicals. LD50 focuses on oral administration of given component in a rat population, whereas our LNP is aimed to administered intravenously and intraperitoneally. Further, for DSPC, the predictions for similar chemicals and chemicals in training data set vary largely. Even for mutagenicity and developmental toxicity, we can expect experimental data to vary from the predicted values.
Further, another obvious issue is the fact that individual components and assembled LNPs with mRNA have variations in toxicity that are difficult to predict by the analysis of individual components only. We also were unable to find other reliable in silico methods to determine immunogenicity of our mRNA-LNP, while leaning over existing literature.
References:
- Morozov V, Rodrigues CHM, Ascher DB. CSM-Toxin: A Web-Server for Predicting Protein Toxicity. Pharmaceutics. 2023 Jan 28;15(2):431. doi: 10.3390/pharmaceutics15020431. PMID: 36839752; PMCID: PMC9966851.
- Gadaleta, D., Vukovic;, K., Toma, C., Lavado, G. J., Karmaus, A. L., Mansouri, K., Kleinstreuer, N. C., Benfenati, E., & Roncaglioni, A. (2019, August 30). Sar and QSAR modeling of a large collection of LD50 rat acute oral toxicity data – journal of Cheminformatics. BioMed Central. https://jcheminf.biomedcentral.com/articles/10.1186/s13321-019-0383-2
- https://biopharma.labcorp.com/catalog/crop-chemical/us-epa-toxicity-estimation-software-suite-test.html
- https://www.epa.gov/chemical-research/toxicity-estimation-software-tool-test
Next, we will look at the Experiments and the results.